Page Contents
WHAT IS IT?
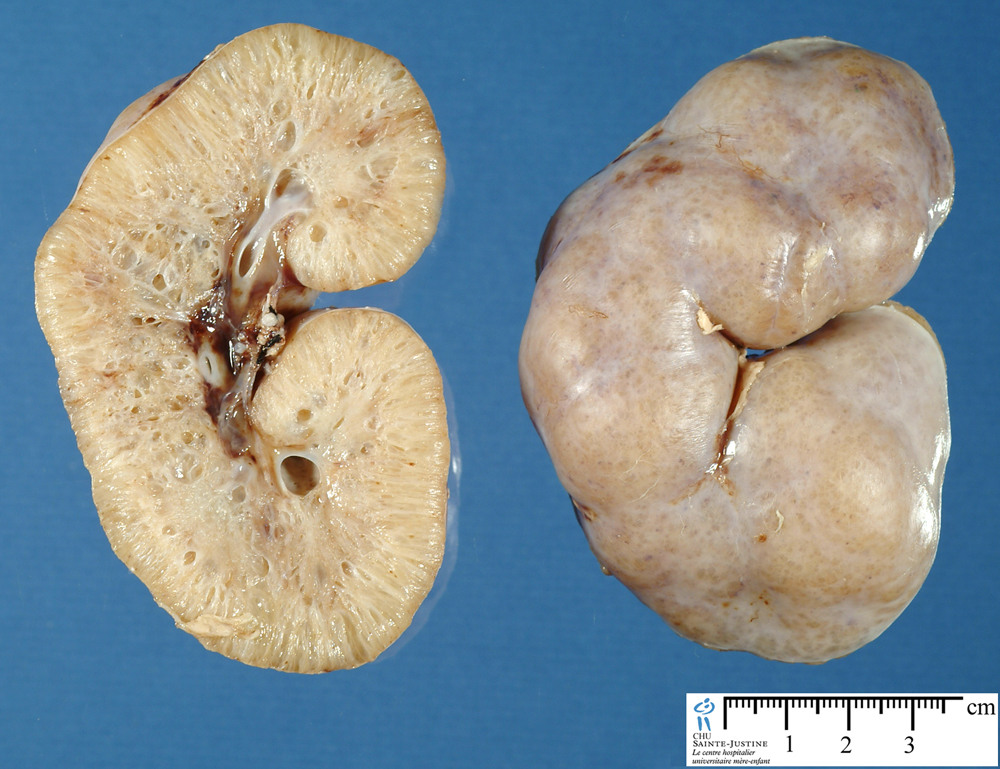
Autosomal recessive polycystic kidney disease (ARPKD) is an autosomal recessive genetic condition that causes many cysts to form on the kidneys in utero. Caused by a mutation on the PKHD1 gene. This usually presents in neonates.
WHY IS IT A PROBLEM?
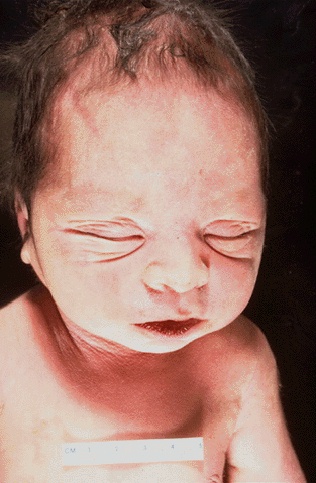
The formation of cysts in the kidneys can cause oliguric renal failure in utero, leading to the lack of production of amniotic fluid (normally the kidneys of the fetus are responsible for generating amniotic fluid). In the absence of this fluid (which provides protective cushion to the fetus against trauma) fetuses will present with Potter’s sequence: facial abnormalities, pulmonary hypoplasia (decreased growth factors preset), and renal insufficiency. ARPKD is usually fatal for many of these reasons.
This condition ultimately not only causes the formation of polycystic kidneys, but also is associated with congenital hepatic fibrosis. After the neonatal period (due to kidney damage and hepatic fibrosis) patients can experience hypertension, renal insufficiency, and portal hypertension.
WHAT MAKES US SUSPECT IT?
Risk factors:
(ARPKD) often will be suspected during prenatal ultrasound screen:
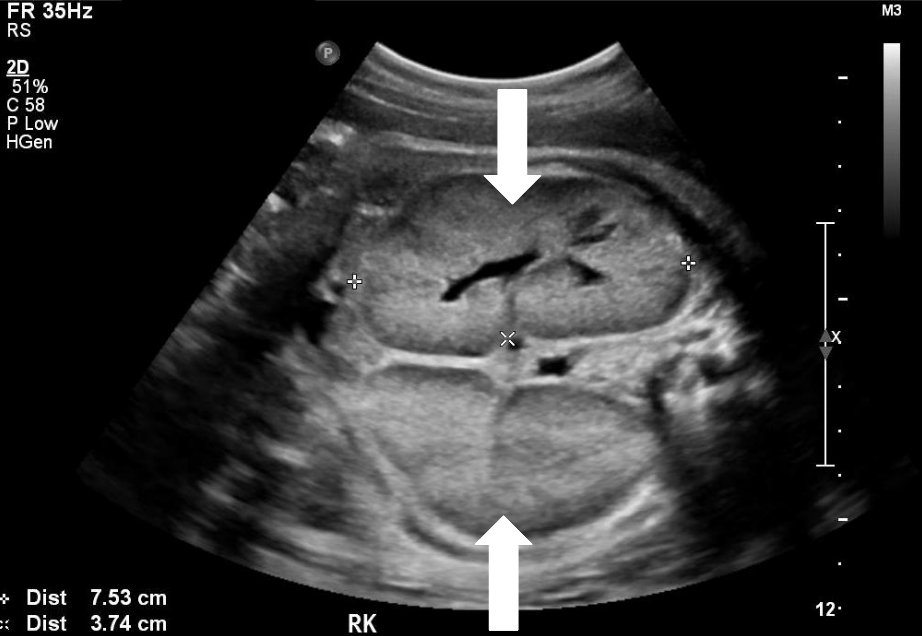
- Bilaterally enlarged echogenic kidneys that also posses poor corticomedullary differentiation
- Small/nonvisualized bladder without any urine
- Oligohydramnios (deficiency of amniotic fluid)
- Reduced lung volume
Potter’s sequence: some patients with ARPKD are born with this condition which refers to the typical physical appearance and associated pulmonary hypoplasia of a neonate, directly resulting from oligohydramnios and compression while in utero.
Bilateral abdominal masses may be present upon conducting physical examination.
HOW DO WE CONFIRM A DIAGNOSIS?
Diagnosis can be confirmed with either pre or postnatal renal ultrasound.
Genetic testing ultimately diagnostic
*Conduct urine analysis to check extent of renal dysfunction
HOW DO WE TREAT IT?
Level IV neonatal intensive care unit recommended for delivery of a child with ARPKD
Treatment can not correct the underlying pathology but instead is focused on addressing symptoms:
- Respiratory support for patients with difficulty breathing
- Nephrectomy for removal of kidneys that are so enlarged they interfere with breathing/feeding
- Electrolyte replacement
- Diuretics/ fluid restriction to avoid fluid overload
- Dialysis
- Kidney transplant
- Liver transplant may also be needed
HOW WELL DO THE PATIENTS DO?
A large fraction of patients die before birth. Many patients die within the first few years of birth. Those that survive respiratory complications often succumb to hepatic complications.
WAS THERE A WAY TO PREVENT IT?
This genetic condition is not preventible.
WHAT ELSE ARE WE WORRIED ABOUT?
Respiratory complications caused by hypoplasia of the lungs is a large contributor to patient mortality
Congenital hepatic fibrosis that can ultimately cause portal hypertension.
OTHER HY FACTS?
Formerly called infantile polycystic kidney disease
FURTHER READING
Page Updated: 02.10.2016